We that observe that a pairwise alignment of two sequences r and s of length n has m mismatches (no gaps).
| |
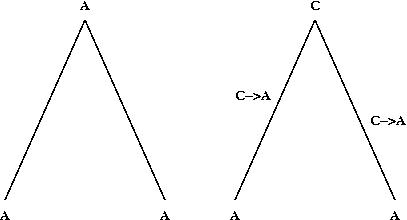
We suspect that this is an underestimate of the actual number of substitutions that occurred. For example, if there is an A at the same position in both sequences, it could be because the ancestral state was also A and no change occured (left hand figure) or because parallel changes occured in both sequences (right hand figure).
 |
Given the observed number of mismatches, we wish to estimate the expected number of substitutions that actually occurred. Let p= m/n be the observed frequency of mismatches. Then, p is an estimator for the expected number of sites at which at least one substitution occurred.
Assume a constant rate of substitution, λ, in both lineages. Let t be the elapsed time since s and r diverged from a common ancestor. The number of substitutions per site is 2λt, which is unknown.
We define a Markov model of instantaneous change. The simplest such model for DNA is the Jukes-Cantor model, which assumes that
The consequences of this assumption are that
To: A G C T
---------------------------------
A | 1-3a a a a
From: G | a 1-3a a a
C | a a 1-3a a
T | a a a 1-3a
First, we define a Markov model of sequence evolution, such as the Jukes Cantor model introduced above.
Second, using this Markov chain we derive an expression describing how changes accumulate over a period of time t. The probability that residue X is in site k after time t is
- 4 a t
pXX(t) = 1/4 + 3/4 e
if we started out in state X (pX(0) = 1) and - 4 a t
pYX(t) = 1/4 - 1/4 e
if we did not start out in state X (pX(0) = 0).
Here X can be A, C, G or T and Y is a nucleotide
different from X.
Third, using the above formulae, we derive an expression for Pmismatch, the expected number of observable differences, between the two sequences:
Pmismatch = 3/4 ( 1 - e - 8 a t).
We solve the above equation for to obtain an expression for a t in terms of Pmismatch:
a t = -1/8 ln ( 1 - 4/3 p ).
Fourth, the expected number of substitutions per site (whether they are observable or not) is 6 a t (see above). Multiplying both sides of the equation by 6, we obtain an expression for the expected number of substitutions per site, in terms of the number of sites with an observable difference:
E[sub] = - 3/4 ln ( 1 - 4/3 Pmismatch ).If we estimate the expected number of observable differences by the number of differences actually observed, m/n, then
E[sub] = - 3/4 ln ( 1 - 4/3 m/n ).So, for example, if we observe mismatches at 10% of the sites, then the Jukes-Cantor model predicts that the actual number of substitutions per site is 0.107.
For more details, see Mona Singh's Phylogeny notes or Durbin, et al: 8.1, 8.2.