Back to "Biological Language Modeling Seminar Topics"
Reference: Ref_LangauerI.html Chapter 7
Protein-ligand docking
= first step in a drug design project: find a lead structure (=small molecule which binds to a given target)
docking problem - predicting the energetically most favorable complex between a protein and a putative drug molecule
if protein structure is know, one can apply docking algorithms to virtually search through the space
2 questions:
1. what does the protein-ligand complex look like
2. what is the affinity with respect to other candidates?
what makes the docking problem hard to solve?
1. scoring problem
= calculating binding affinity given a protein-ligand complex
- no general scoring function is available
2. large number of degrees of freedom
- most important degrees of freedom:
1. relative orientation of the two molecules
2. conformation of the ligand
3. protein conformation
4. water molecules can be between ligand and protein
5. protonation state
Types of docking problems
= two macromolecules are docked, such as protein and DNA, or protein and protein
- large contact area
- molecules have fixed overall shape
=> methods based on geometric properties like shape complementarities alone can be efficiently used to create energetically favorable complexes
2. Small molecule docking
= a small molecule is docked to a macromolecule
- ligand is typically not fixed in shape (as opposed to macromolecular docking)
- typical ligand size has 5-12 rotatable bonds
- often fragments of ligand are used for modeling, eg. combinatorial libraries are docked by combining placement for individual building blocks of the library
Differences in protein-ligand and DNA-ligand docking:
- DNA specific binding mechanisms:
1. covalent binding
2. binding of intercalators (= molecule that binds sandwiched between two bases)
- DNA undergoes conformational changes upon binding - need to take DNA flexibility into account
1. by docking into distorted DNA structure
2. by taking DNA dynamics into account, e.g. (Zacharias and Sklenar (1999) J. Computational Chemistry 20, 287-300) based on normal mode analysis for handling structural changes during docking calculations
Steps:
1. Find a set of compounds to start with
- e.g. from inspecting known ligands for a protein (e.g. substrate in an enzyme)
2. compounds from a screening experiment of a combinatorial library (in which there is usually a molecular fragment that is common between all molecules of the library, the core, and the fragments attached to the core are R-groups)
3. compounds from a filtering experiment using other software
4. from varying other lead structures or known ligands
5. virtual screening using a fast docking algorithm (typically from a million molecules)
6. de novo design using fragments of compounds
=> get several hundred to thousands of ligands to start with
Docking Methods:
I. Rigid-body docking algorithms
- Historically the first approaches.
- Protein and ligand are held fixed in conformational space which reduces the problem to the search for the relative orientation fo the two molecules with lowest energy.
- All rigid-body docking methods have in common that superposition of point sets is a fundamental sub-problem that has to be solved efficiently:
Superposition of point sets:
minimize the RMSD, see section 7.2.1.4. in Lengauer page 323
Ia. Clique-search based approaches
= matching characteristic features of the two molecules (Kuhl, Crippen, Friesen (1984) J. Computational Chemistry 5, 24-34)
use a graph to search for compatible matches: the vertices of the graph are all possible matches and edges connect pairs of vertices representing compatible matches
compatibility = distance compatibility with in a fixed tolerance epsilon
The matches (p1,l1), (p2,l2) are distance-compatible if |d(p1,p2)-d(l1,l2)|<epsilon
Example: DOCK program (see links)
today most widely used molecular docking program
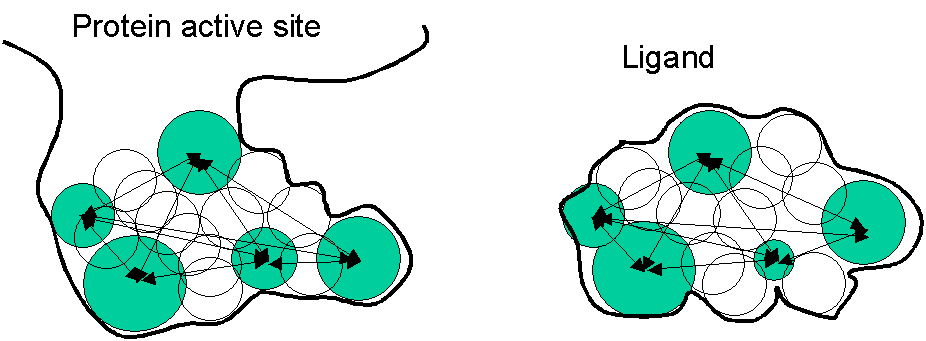
- starting with the molecular surface of the protein , a set of spheres is created inside the active sties
- the spheres represent the volume which could be occupied by the ligand: VOLUME is the feature used for matching
- ligand is represented by spheres inside the ligand (see figure)
http://www.cmpharm.ucsf.edu/kuntz/dock_demo.html
Figure: DOCK program works like this: match the spheres shown in green (distances between them are used for scoring).
Extensions:
- matching spheres can be labeled with chemical properties
- use distance bins to speed up the process
- use ensemble of protein structures (takes into account limited protein flexibility, see below)
- use chemical interactions as the feature for matching instead of volume, e.g. ADAM (uses H-bonds)
- LIDO (de novo ligand design program) matches H-bond vectors and hydrophobic points
- CLIX takes energetically favorable regions for functional groups of the ligand as a model. These regions are pre-calculated with program GRID (see below). First take two points for initial matching, and then rotate in regular intervals).
Ib. Geometric hashing
originates from computer vision and was first applied to molecular docking program sby Fischer, Norel Nussinov Wolfson (1993) CPM93, 20-34 and Fischer, Lin, Wolfson and Nussinov (1995) J. Mol. Biology 248, 459-477.
Given a picture of a scene and a set of objects within the picture, both represented by points in 2d space, the goal is to recognized some of the models in the scene
1. preprocessing phase: create a hash table from the set of models (for each model, each pair of points define a basis)
2. for each basis, every third point that belongs to the model is expressed in coordinates relative to the basis
3. store a tuple(model, basis) in a has table addressed by teh relative coordinates of the third point
(this separate table is necessary, because it is possible that the third point is occluded in the scene).
Matching:
create a (model,basis) table in the same way as above for the query
match to original table
advantages:
time-efficient
deals with partial matching (partially occluded objects( - this is important because usually not all the features of a ligand are in contact with the protein)
Specifics:
In order to apply geometric hashing to molecular docking, us the sphere representation of DOCK as the underlying model
three points to define a axis
number of has table entries increases with the 4th power of the number of ligand atoms
therefore the basis is described by only two points leaving one degree of freedom open (rotation around the axis defined by the two points).
Ic. Pose clustering
originally developed to detect objects in pictures with an unknown camera location
algorithm matches each triangle of object points in the two pictures
from a match, calculate the position of the camera, and store the locations and cluster them
identify large clusters
LUDI model is used as underlying model of molecular interactions (see Figure 7.3): interaction points are approximated by discrete points. (More details see book)
II. Flexible ligand docking algorithms
most ligands have large conformational spaces with several low energy states
IIa. Conformation ensembles of rigid bodies
this uses the same algorithms as the rigid-body docking algorithms, but apply them to ensembles of rigid structures, each representing a different conformation of the ligand
Flexibase/FLOG algorithm:
- store a small set of diverse conformations for each molecule from a given database created with distance-geometry methods
- select 25 conformations based on dissimilarity criteria (RMSD)
Lorber and Shoichet approach:
- generate 300 conformations per molecule, and define one rigid part of the molecule and superimpose
- faster than independent docking
IIb. Flexible docking based on fragmentation
ligand is divided into several fragments.
Each fragment is either rigid or has a small number of conformations
- place and join algorithms
multiple fragments are all docked independently
then, placements for adjacent fragments in which there is overlap are identified and reconnected
works well if the molecule consists of a small set of medium-sized rigid fragments
if the fragments are to small they cannot be placed independently
difficult to generate correct bond lengths and angels at the connecting atoms without destroying the interactions of the fragments to the protein
- incremental construction algorithms
more frequently used than place and join
place one fragment, the base of anchor fragment, in the active site
details see Lengauer
IIc. Genetic algorithms and evolutionary programming
genetic algorithm is a general purpose algorithm to optimize something by mimicking the process of evolution
fitness function is used to decide which individuals (= configurations in the search space) survive and produce offspring
Requirements:
1. Design a description of a configuration that describes all degrees of freedom of the problem without redundancy and models constrains of the configuration space directly so that configurations violating the constrains are never generated ruing the optimization "the chromosome"
2. develop a fitness function
similar to scoring functions
3. optimization scheme needs to chose population size, the number of generations, crossover, and mutation rates etc.
optimize fitness function and keep the diversity in the population
GOLD:
- stores two things in the chromosome:
all torsion angles
all H-bonds
IId. Distance geometry
- comes from NMR
- instead of describing a molecule by coordinates in Euclidean space, it is described by a distance matrix, containing all interatomic distances
based on distance matrices, a set of conformations can be described in a comprehensive form by calculating a distance interval for each atom pair
- uses clique search and distance compatibility: two matches between protein and ligand atoms are compatible if the site point distances lies within the distance interval of the ligand atom pair
- drawback: the distance matrix is over-determined
IIe. Random search
LIGIN:
- generate a large set of starting conformations radnomly
- optimize by surface complementarity adn H-bond geometry
PRO_LEADS:
- tabu search reduces the number of similar structures in the random set by avoiding them (the entry in the tabu list is only replaced if it has a better scoring function)
III. Docking by simulation
= instead of enumerating a discrete low-energy subspace of the problem, begin the calculation with a starting configuration and locally move to configurations with lower energy
IIIa. Simulated annealing
1. start with configuration A with an energy or score value E(A)
2. then, random local move to a new configuration B weith energy E(B)
3. accept the new configuration based on the Metropolis criterion where E(B)<E(A) or with probability P = exp(-(E(B)-E(A))/kT)
4. over simulation time, reduce the temperature T based on a cooling schedule such that it accepts configuration with increased energy becomes less likely
AUTODOCK and others
IIIb. Molecular dynamics
1. a force-field is used to calculate the forces on each atom of the simulated system
2. follow Newton mechanism, calculate velocities and accelerations and the atoms are moved slightly with respect to a given time step
- the simulation becomes more exact as the time step is decreases and the more atoms of the system are taken into account, but this is too time-consuming and therefore unpractical
- various methods to decrease computation time (see Lengauer)
IIIc. Monte-Carlo algorithms
in MD, local movements are determined by force field
in Monte-Carlo, they are random
(simulated annealing is an example for a Monte-Carlo method)
IIId. Hybrid models
fragment-based approaches and genetic algorithms are good for wide coverage of onfiguration space, but simulation-based methods otuperform in finding exact local minimal
hybrid models have one of each step in it
IV. Docking of combinatorial libraries
has special needs, see details Lengauer 7.2.4.
Scoring methods:
given a protein and a ligand in aqueous solution what is the ligand's free energy of binding to the protein?
Ranking task during a docking calculation: different orientations of the same ligand in the same active site have to be evaluation and the scoring function has to distinguish them
problems:
- assumption that free energy of binding is dominated by a single mode
- solvation effects difficult to quantify
- conformational changes of protein difficult to quantify
- loss of entropy during complex formation difficult to quantify
Types of scoring functions in use:
1. force-field scoring
- (Lennard Jones or other potentials), problem does not take entropic and
solvation effects into account. Example GRID.
2. empirical scoring
- correlation of geometric parameters to measured free binding energies
sum of terms representing different physico-chemical effects contributing to the binding energy (incl. H-bonds, hydrophobic contacts etc.)
3. knowledge-based (or PMF, potentials of mean force)
- assumption: situations frequently seen in structures are energetically favorable (used also in structure prediction)
- based on inter-atom distances
- for each atom type pair, the number of occurrences are counted depending on the distance
- convert the resulting histogram into an energy function
score = add the energy function values for all inter-atom pairs of the complex
- add a solvent-accessible surface term to take solvation into account
- add a volume correction factor to account for the ligand volume around each ligand atom which avoids interactions with the protein
References 143-148 in Lengauer
Validation studies
1. Reproducing X-ray crystal structures
2. validated blind predictions
3. screening small molecule databases
4. docking combinatorial libraries
Molecular docking in practice:
1. Preparing input data
2. Analyzing docking results
3. Choosing the right docking tool